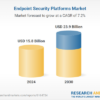






























A pioneering study led by researchers Weihong Tan, Xiaohong Fang, and Tao Bing from the Hangzhou Institute of Medical Sciences at the Chinese Academy of Sciences has introduced a machine learning-based approach to nucleic acid aptamer discovery, significantly enhancing the speed and accuracy of this crucial area of research. Their work, published in CCS Chemistry on January 7, 2026, demonstrates an innovative framework that decodes complex secondary structures of nucleic acid aptamers from single-round screening data, bypassing the traditionally lengthy iterative enrichment processes.
Nucleic acid aptamers, which are short, single-stranded oligonucleotides, are valued for their ability to bind with high specificity to various target molecules. The conventional method known as SELEX (Systematic Evolution of Ligands by EXponential enrichment) has historically been employed to generate candidate aptamers. However, determining the functional secondary structures that enable target recognition remains a significant challenge. Traditional structural methods like electron microscopy, nuclear magnetic resonance (NMR), and X-ray crystallography are resource-intensive and often fail to resolve the dynamic and heterogeneous structures of aptamer-target complexes, limiting the optimization of aptamer sequences.
In response to these challenges, the research team developed a sophisticated machine learning framework that combines unsupervised autoencoder clustering with deep learning algorithms to analyze the core sequence elements within large pools of aptamer candidates obtained from a single screening round. This methodology enables the identification of conserved sequence motifs and their corresponding secondary structural features critical for target binding, allowing for the computational inference of the molecular architecture underlying aptamer function. This marks a significant departure from traditional methods, which rely heavily on extensive experimental trial-and-error.
The researchers applied their technique to a dataset of aptamers targeting the CD8 protein, a key cell surface marker. By employing deep learning to analyze sequence families within the single-round library, they identified a prominent core sequence, “GTGAGGAGCTTGAAA,” amidst a highly heterogeneous background. This finding highlighted the advantages of machine learning over conventional multiple sequence alignment methods, which struggled to extract short motifs from low-homology environments.
To validate their computational findings, the team synthesized a candidate library embedding the critical partial motif (5′-AGCTTGAAA-3′) and subjected it to RE-SILEX, a selection method. Remarkably, all newly identified aptamers—over 20,000 in total—contained the predicted core sequences, confirming the robustness of their machine learning-enhanced screening process. This not only demonstrated the identification of biologically relevant motifs but also facilitated subsequent aptamer enrichment and design.
Further analysis revealed that approximately 62.4% of these sequences formed stem-loop structures essential for molecular recognition, with the sequence “GTGA” frequently found in multi-branched loops and stem regions, suggesting a consensus binding motif. Detailed quantification confirmed a shared secondary structure among the aptamers, illustrating how specific folding patterns confer target specificity and affinity. Informed by these insights, the researchers implemented rational truncation and optimization strategies, which significantly enhanced binding affinities by eliminating redundant nucleotides and concentrating on functionally critical motifs.
The versatility of this machine learning technique was further illustrated through its application to a different fibrogenic target, the fibroblast activation protein (FAP). The method identified a highly conserved core sequence believed to form a G-quadruplex secondary structure, demonstrating the framework’s adaptability to complex, non-canonical nucleic acid topologies. Subsequent optimization of FAP aptamers further validated the broad applicability of this novel approach.
In follow-up validation experiments focusing on the CD8 protein, the researchers found that over three-quarters of sequences contained the identified core motif and shared coherent secondary structural elements. The truncated and optimized aptamers exhibited affinity enhancements exceeding tenfold, while maintaining target specificity within complex cell environments. Notably, these structural insights also paved the way for the design of split-type aptamers and new sequence generation, marking significant advancements in synthetic biology and functional nucleic acid design.
This research signifies a transformative shift in aptamer discovery, moving from iterative enrichment and experimental structural elucidation to data-driven, computational methodologies. The findings challenge the conventional belief that extensive screening cycles are necessary for effective aptamer identification. By revealing that single-round libraries contain rich structural and functional information, the study emphasizes the role of high-throughput sequencing coupled with advanced machine learning in rapidly decoding the nucleic acid folding landscapes governing target binding.
The implications of this work extend beyond aptamers, suggesting potential applications in broader biomolecular and therapeutic contexts. The machine learning algorithms developed can be adapted for the exploration of non-coding RNA interactions and RNA-protein complex modeling, enabling AI-driven virtual screening platforms for nucleic acid ligands. This advancement is expected to accelerate drug discovery pipelines and enhance precision diagnostics, paving the way for next-generation nucleic acid-based therapeutics tailored for personalized medicine.
Supported by various funding agencies, including the National Natural Science Foundation of China and the Zhejiang Provincial R&D Program, this research exemplifies the powerful intersection of computational innovation and molecular biology. The publication in CCS Chemistry highlights the increasing importance of interdisciplinary methodologies in advancing chemical sciences globally.
See also UCT Researcher Dr Tim Köhler Wins Global Grant to Study AI’s Impact on Youth Unemployment
UCT Researcher Dr Tim Köhler Wins Global Grant to Study AI’s Impact on Youth Unemployment MIT Develops AI System to Analyze Figure Skating Jumps, Enhancing Performance Metrics
MIT Develops AI System to Analyze Figure Skating Jumps, Enhancing Performance Metrics Harvard Study Reveals Employees Work Harder with AI, Risking Burnout and Fatigue
Harvard Study Reveals Employees Work Harder with AI, Risking Burnout and Fatigue ByteDance Launches Seedance 2.0, Revolutionizing AI Video Creation with Multimodal Inputs
ByteDance Launches Seedance 2.0, Revolutionizing AI Video Creation with Multimodal Inputs